Вероятно, самый простой способ обойти эту проблему - это напрямую изменить код для функции Биоинформатика Toolbox SEQLOGO (если это возможно).В R2010b вы можете сделать:
edit seqlogo
И код функции будет показан в редакторе.Затем найдите следующие строки (строки 267-284) и либо закомментируйте их, либо удалите их полностью:
S_before = log2(nSymbols);
freqM(freqM == 0) = 1; % log2(1) = 0
% The uncertainty after the input at each position
S_after = -sum(log2(freqM).*freqM, 1);
if corrError
% The number of sequences correction factor
e_corr = (nSymbols -1)/(2* log(2) * numSeq);
R = S_before - (S_after + e_corr);
else
R = S_before - S_after;
end
nPos = (endPos - startPos) + 1;
for i =1:nPos
wtM(:, i) = wtM(:, i) * R(i);
end
Затем поместите эту строку на их место:
wtM = bsxfun(@times,wtM,log2(nSymbols)./sum(wtM));
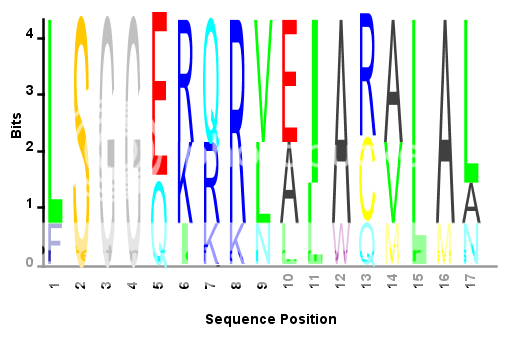
Вы будетеВозможно, вы захотите сохранить файл под новым именем, например seqlogo_norm.m, поэтому вы все еще можете использовать исходную неизмененную функцию SEQLOGO .Теперь вы можете создавать графики профиля последовательности со всеми столбцами, нормированными на одинаковую высоту.Например:
S = {'LSGGQRQRVAIARALAL',... %# Sample amino acid sequence
'LSGGEKQRVAIARALMN',...
'LSGGQIQRVLLARALAA',...
'LSGGERRRLEIACVLAL',...
'FSGGEKKKNELWQMLAL',...
'LSGGERRRLEIACVLAL'};
seqlogo_norm(S,'alphabet','aa'); %# Use the modified SEQLOGO function
СТАРЫЙ ОТВЕТ:
Я не уверен, как преобразовать информацию профиля профиля, чтобы получитьжелаемый вывод из функции набора инструментов биоинформатики SEQLOGO , но я могу показать вам, как изменить альтернативу seqlogo_new.m, которую я написал для моего ответа на связанный вопрос , с которым вы связались.Если вы измените строку, которая инициализирует bitValues, с этого значения:
bitValues = W{2};
на следующее:
bitValues = bsxfun(@rdivide,W{2},sum(W{2}));
Затем вы должны масштабировать каждый столбец до высоты 1. Например:
S = {'ATTATAGCAAACTA',... %# Sample sequence
'AACATGCCAAAGTA',...
'ATCATGCAAAAGGA'};
seqlogo_new(S); %# After applying the above modification
